
Knowledge of how cells and organisms defend themselves against pathogenic microbes is of paramount interest in our understanding of infection and immunity. Determining the mechanism by which cells achieve this – and exploiting this knowledge – may aid in the development of therapies as well as improving our understanding of how these microbes cause disease.
Shu et al., (from the lab of Carolyn Coyne at the University of Pittsburgh) recently published the finding that the human gene ADAP2 (also known as centaurin alpha 2) helps in the defence against two RNA viruses (dengue virus and vesicular stomatitis virus – two arboviruses) by inhibiting their entry into cells. Focusing on trying to find genes that can affect entry, the Pittsburgh team used gene over-expression, live cell fluorescence microscopy and bespoke virus entry assays, to characterise the biology of the ADAP2 protein – of which, like many genes, very little is known about. Through doing so they extend our understanding of the human antiviral response. But how did they make their discovery?
Why target virus entry?
Viruses are obligate intracellular parasites and so need to get inside cells in order to replicate, make new copies of themselves and spread infection. The major barrier to this is the eukaryotic cell plasma membrane, a hydrophobic lipid bilayer, impermeable to the large virus particles. Virus entry can be a highly co-ordinated and complex process: while some viruses can enter directly at the cell surface, others appear to trick the cell into bringing them inside the cell inside small vesicles where they can eventually break free and begin replication.
Influenza virus has a well-studied and exemplary entry pathway. In its simplest depiction: influenza virus particles bind to cell surface sialic acids (a kind of sugar molecule), signalling to the cell to begin a process called endocytosis, engulfing the virus particle in a membrane and pulling this structure into the cell with the cytoskeleton. Through the journey inside the cell, the vesicle’s pH is eventually decreased, triggering the viral surface proteins to extend and dig into its entrapping vesicle membrane causing fusion of the virus and cell membranes, releasing the virus genome into the cell primed for replication.

Many other viruses carry out very similar processes and our cells have adapted to how viruses gain entry and have evolved intricate means to prevent this from happening. For example, one family of genes, the IFITM genes and their encoded proteins (such as IFITM1), can potently block virus entry (in particular, membrane fusion). Blocking entry can be an effective and early means to control an infection. These genes, and other genes, are regulated by the antiviral signalling molecule: interferon and are termed ‘ISGs’, or interferon-stimulated genes. These ISGs can play a very important role in defending us against virus infection.
ADAP2 can inhibit dengue and vesicular stomatitis virus entry
Shu et al., set out to determine whether human cells had any other genes that could prevent virus entry independent of the IFITM family. ADAP2 was found to be an ISG, a group of hundreds of genes that are expressed in response to interferon signalling, which trigger an antiviral environment within a cell, following IFN beta treatment of HT1080 cells, a commonly used cancer cell line.
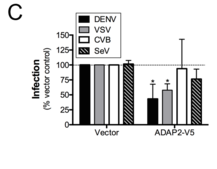
ADAP2 was thought to have the potential to affect entry because it has Arf GAP activity (i.e aids hydrolysis of GTP to GDP-bound to Arf proteins, which alters Arf protein function) associated with other proteins that regulate entry and is known to associate with the actin cytoskeleton. These both are necessary for the complex mode of virus entry. Over-expression of ADAP2 even inhibited infection of 293T cells with dengue virus (DENV) and vesicular stomatitis virus (VSV), but not Coxsackie B virus nor Sendai virus.
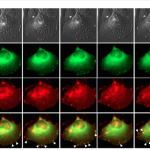
Overexpression of ADAP2 did not lead to expression of other ISGs, suggesting a direct mechanism of antiviral activity. Live-cell confocal microscopy analysis of cells overexpressing a fluorescent protein-tagged ADAP2 alongside fluorescent protein-tagged actin showed that it dramatically altered cytoskeletal structures, inducing plasma membrane ‘ruffling’; changes in actin localisation and induced macropinocytosis. Further evidence for its role in macropinocytosis came from the observation that ADAP2 localised with Rab34-associated vesicles. It also localised with recycling endosomes, which eventually associated with lysosomes, the places where the cell can digest contents.
Induction of macropinocytosis as well as its antiviral activity was dependant of its Arf GAP activity. In particular ADAP2 has GAP activity for the protein Arf6, a protein involved in regulation of endocytosis. This suggests a model where by ADAP2 induces a process similar to macropinocytosis via its regulation of Arf6 that causes the endocytosis of viruses, leading to their transport to – and degradation in – host lysosomes. Importantly this process competes with the standard viral entry pathway thus inhibiting productive infection.
What does ADAP2 really do?
This paper from the Coyne lab presents data suggesting that ADAP2 functions to inhibit infection with two viruses, very likely through altering their entry pathways, and is a fascinating study into the cell biology of an ISG.

However, the role of ADAP2 during a complete interferon response was barely addressed in this study. In fact, the authors report that knockdown of ADAP2 led to an increase in dengue virus infection. While they show that ADAP2 CAN inhibit some infections, it is not clear whether this is its main role and whether or not the promotion of macropinocytosis is a consequence – and an artefact – of its over-expression in lab cell lines. This question could be answered by using loss of function and rescue of expression using something like RNAi or gene knockout approaches in a cell line that expresses relevant levels of ADAP2 and can be infected with the viruses studied.
This is especially important as other infection related processes may be affected by promotion of macropinocytosis and some viruses, such as the Ebola virus, can productively infect cells via macropinocytosis. Promotion of this process might thus increase the likelihood of infection.
If ADAP2 is important, it might be expected that viruses have evolved mechanisms to evade it. Many viruses, such as hepatitis C virus, go to great lengths to fine-tune their entry into cells in order to promote the best possible chance of kicking off a productive infection. It is quite possible that even under these over-expressing conditions, dengue and VSV have evolved means to circumvent the inhibitory effects of interferon-mediated changes in entry induced by genes like ADAP2, that could reduce the entry inhibition caused by its over-expression.
If it turns out that ADAP2 does not actually inhibit infection under physiologically relevant conditions, it does suggest that manipulation of virus entry via cytoskeletal rearrangements by promoting macropinocytosis (existing inhibitors) could be a good strategy to inhibit these infections. Is it speculative whether we could manipulate virus entry through small molecule drugs? Or maybe evolution has led to other organisms using ADAP2-like genes to inhibit infection? However, why human ADAP2 would do this already is unknown; maybe inhibitory levels of ADAP2 activity is mutually exclusive to a healthy cell. Other studies have shown that it can also directly stabilise microtubules and promotes healthy neuronal and heart development.
Final remarks
In conclusion this study provides evidence that ADAP2 is a human ISG that can inhibit virus infection by altering virus entry, thus extending our understanding of the interferon response. Although it would be fascinating to put ADAP2 activity into context of a whole interferon response.
By Connor Bamford Ph.D @cggbamford
The CVR has a broad collaborative programme in host innate and intrinsic immunity, and owing to its relevance, even bridges many of our research themes. In particular, the CVR investigates the host cell biology of these anti-viral genes; how these genes affect virus replication; how viruses have evolved to inhibit their action; and what role do these genes play in virus pathogenesis and in the clinic. These projects are explored across diverse viruses (bluetongue, bunyaviruses, lentiviruses, herpes viruses, arboviruses, influenza, hepatitis C virus), hosts (human, other mammalians and arthropods) and groups, such as those of Massimo Palmarini, Sam Wilson, Richard Elliott, Chris Boutell, Roger Everett, Alain Kohl, Pablo Murcia and John McLauchlan.